

Pivotal trial study design1
The efficacy and safety of dimethyl fumarate were demonstrated in 2 studies (Study 1 and Study 2), both of which enrolled patients with RRMS who had experienced at least 1 relapse over the year preceding the trial or had an MRI scan demonstrating at least 1 gadolinium-enhancing lesion within 6 weeks prior to randomization.
Study 1: a 2-year, randomized, double-blind, placebo-controlled study in which 1234 patients with RRMS were randomized to receive dimethyl fumarate 240 mg BID (n=410), dimethyl fumarate 240 mg TID (n=416), or placebo (n=408).
Study 2: a 2-year, randomized, double-blind, placebo-controlled study in which 1417 patients with RRMS were randomized to receive dimethyl fumarate 240 mg BID (n=359), dimethyl fumarate 240 mg TID (n=345), an open-label comparator (n=350), or placebo (n=363).
The only approved dose of dimethyl fumarate is the 240 mg BID dose.
The adverse reactions presented in the table below are based on safety information from 769 patients treated with dimethyl fumarate 240 mg twice a day and 771 placebo-treated patients.
| DMF n=769 |
Placebo n=771 |
|
|---|---|---|
| Flushing | 40% | 6% |
| Abdominal pain | 18% | 10% |
| Diarrhea | 14% | 11% |
| Nausea | 12% | 9% |
| Vomiting | 9% | 5% |
| Pruritus | 8% | 4% |
| Rash | 8% | 3% |
| Albumin urine present | 6% | 4% |
| Erythema | 5% | 1% |
| Dyspepsia | 5% | 3% |
| Aspartate aminotransferase increased | 4% | 2% |
| Lymphopenia | 2% | <1% |
Study was funded by Biogen
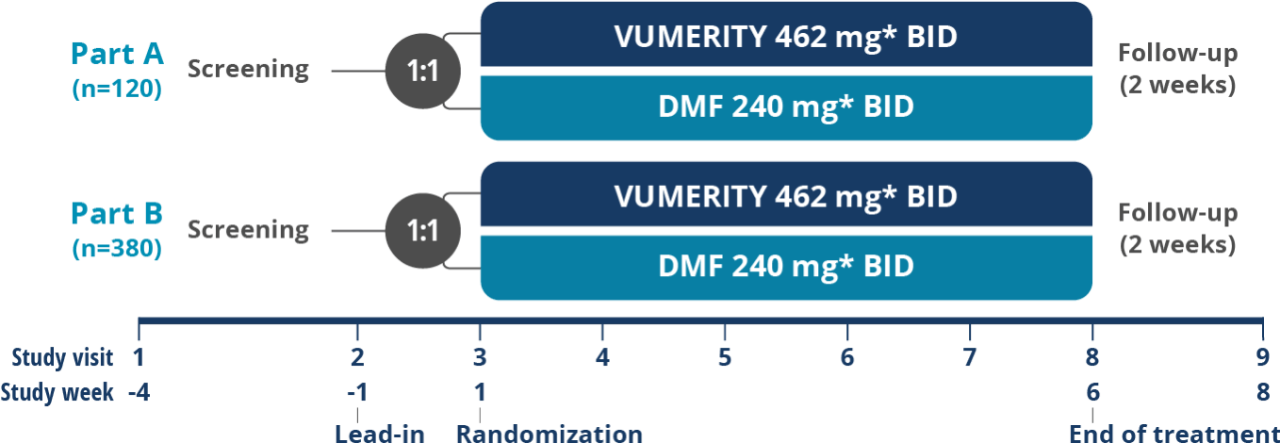
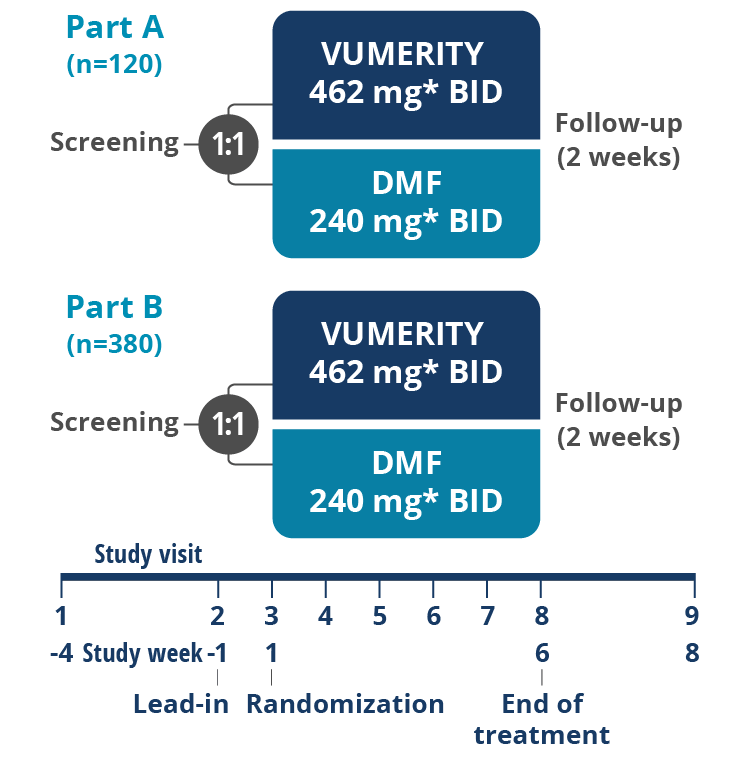

*In study week 1, the titrated dose for VUMERITY was one 231 mg delayed-release capsule taken orally, twice daily. The titrated dose for dimethyl fumarate was one 120 mg delayed-release capsule taken orally, twice daily. From study weeks 2–5, the full dose for VUMERITY was two 231 mg delayed-release capsules taken orally, twice daily, and the full dose for dimethyl fumarate was one 240 mg delayed-release capsule taken orally, twice daily.
Secondary endpoints were assessed in this study, but results are not presented here.
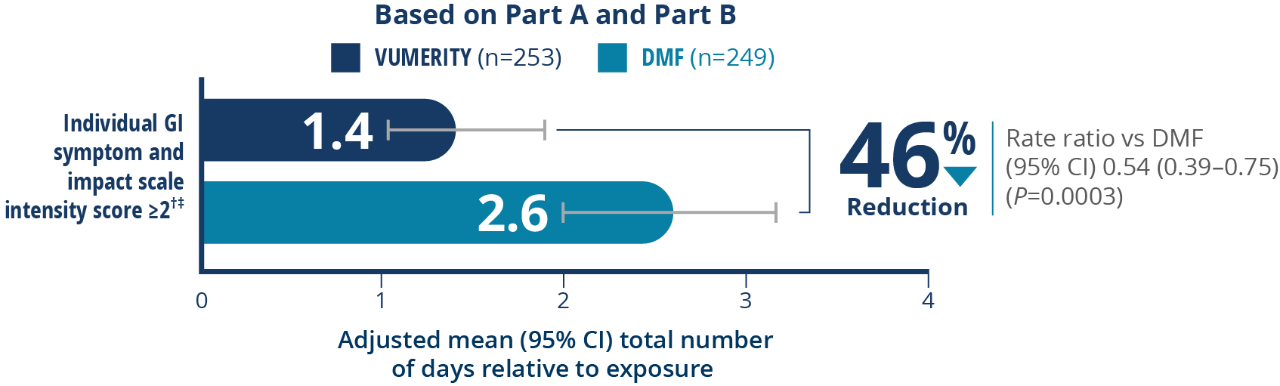
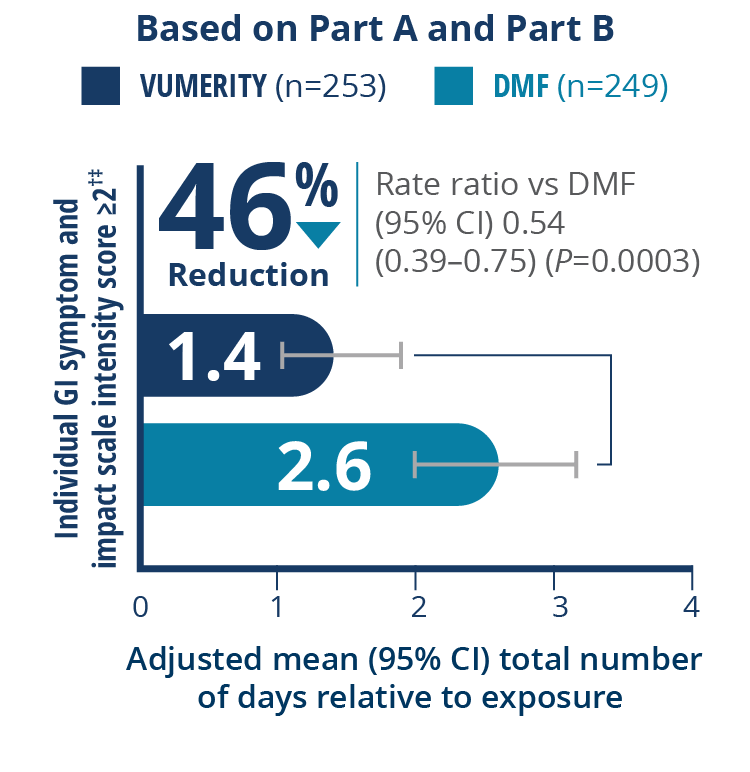

Overall, 19.3% of patients (17 out of 88) in the VUMERITY group and 30.6% of patients (37 out of 121) in the DMF group used concomitant medications to treat GI-related AEs during the treatment period
†Intensity of nausea, vomiting, upper abdominal pain, lower abdominal pain, and diarrhea reported twice per day. ‡Patients completed ≥1 postbaseline tolerability eDiary assessment and were included in the analysis of patient-assessed GI tolerability.
| Patients, n (%) | VUMERITY§ n=254 |
DMF§ n=252 |
|---|---|---|
| Received ≥1 dose of study drug | 253 (99.6) | 251 (99.6) |
| Completed the treatment period | 245 (96.8) | 233 (92.8) |
| Mean (SD) days of treatment exposure, IGISISII | 35.2 (4.2) | 34.2 (5.9) |
| Mean (SD) days of treatment exposure, GGISISII | 33.3 (4.8) | 32.6 (5.6) |
| Rolled over to EVOLVE-MS-1 | 239 (94.5) | 225 (89.6) |
| Discontinued the study | 8 (3.2) | 18 (7.2) |
| AE leading to discontinuation during the treatment period | 4 (1.6) | 14 (5.6) |
| GI AE leading to discontinuation | 2 (0.8) | 12 (4.8) |
| Upper abdominal pain | 0 | 5 (2.0) |
| Diarrhea | 1 (0.4) | 3 (1.2) |
| Abdominal pain | 0 | 3 (1.2) |
| Vomiting | 1 (0.4) | 2 (0.8) |
| Abdominal distension | 0 | 1 (0.4) |
| GI pain | 0 | 1 (0.4) |
| Nausea | 0 | 1 (0.4) |
§Patients were randomized.
‖VUMERITY, n=253; DMF, n=249.
6x
fewer discontinuations due to GI adverse events compared to dimethyl fumarate (0.8% of patients taking VUMERITY vs 4.8% of patients taking DMF)

| System organ class preferred term, n (%) | VUMERITY n=253 |
DMF n=251 |
|---|---|---|
| Any AE | 198 (78.3) | 210 (83.7) |
| GI disorders | 88 (34.8) | 123 (49.0) |
| Diarrhea | 39 (15.4) | 56 (22.3) |
| Nausea | 37 (14.6) | 52 (20.7) |
| Upper abdominal pain | 17 (6.7) | 39 (15.5) |
| Abdominal pain | 16 (6.3) | 24 (9.6) |
| Lower abdominal pain | 15 (5.9) | 17 (6.8) |
| Vomiting | 9 (3.6) | 22 (8.8) |
Study was funded by Biogen